1. Snakebite therapies in Colombia
1.1 What is an antivenom?
Although there is no consensus on the exact definition of an antivenom, it is generally thought to be a drug or treatment that counteracts the harmful effects attributed to a venom or disease [1]. Humans have developed specific antidotes to defend themselves against these substances, including the group of antivenoms, also known as antisera. Purified antibodies (immunoglobulin-based therapies) have historically played a fundamental role in deactivating toxins from specific venoms [2–4].
According to the Colombian regulatory framework for obtaining sanitary registrations described in Decree 386 of 2018 [10], antivenoms are defined as "purified fractions of immunoglobulins or immunoglobulin fragments from plasma obtained from animals immunized with a venom or a mixture of venoms." Antivenoms are prepared by hyperimmunizing animals such as sheep or horses and injecting them with small doses of venom to produce antibodies that deactivate harmful toxins. The antibodies are collected by drawing blood from the animal [3–5]. After the antibodies are isolated, the antivenoms are produced and used to counteract the envenoming caused by snakebites. An antivenom is composed of equine-origin antibodies, which correspond to a purified fraction of hyperimmune sera. This is the only way to save the lives of envenomed victims [5,6,7]. The treatment consists of administering these antibodies that are responsible for neutralizing the venom’s toxins.
Although this antivenom is an essential drug for treating snakebite accidents, especially in tropical and subtropical countries [8], its distribution is unfortunately inadequate. Only a few countries in tropical latitudes meet the adequate quality standards to manufacture these substances. In addition, some countries lack proper regulation and control over their production, and this greatly impedes assessment and intervention in the control of the quality and effectiveness of these antidotes [9].
1.2A brief history of antivenom production in the Colombian National Health Institute
The National Institute of Health (INS) is a governmental scientific and technical entity that has been formally producing polyvalent antivenom serum since the 1970s (see Chapter 7). This serum is developed from hyperimmune plasmas obtained through standardized immunization protocols using venoms from medically important snake species across different ecoregions of Colombia [11].
According to INS production records, the first extractions of equine-derived hyperimmune plasma for antivenom production date back to the early 1970s. Hyperimmune anti-rabies plasma production was also initiated during this period. In February 1972, the INS registered the production of Bothropic and Crotalic hyperimmune plasmas, along with initial trials for antivenom production. At that time, challenges arose due to limited experience in the handling of snake venoms, immunization protocols, titration techniques, and production procedures. The first batch consisted of approximately 150 vials, but production was interrupted due to these difficulties (see Chapter 7).
Two years later, the physicians Augusto Corredor, Miguel Guzman, and Ernesto Barbosa from the INS attended the Tropical Medicine Congress in Medellin. MD. Roger Bolaños, renowned for his extensive experience in antivenom production and snakebite management, also attended the event. The INS physicians contacted MD. Bolaños through the Pan American Health Organization (PAHO) to hire him as a consultant for the INS serum laboratory. MD. Bolaños trained the production team, including Juan Manuel Rengifo, Guiomar Caicedo de Pardo, and Carlos Cáceres, in antivenom production (see Chapter 7). They successfully produced an experimental batch of hyperimmune plasma from horses immunized with Bothrops asper venom, which was approved for its neutralizing capacity. This early success facilitated the development of new production protocols and immunization schemes, solidifying the formal establishment of antivenom production in Colombia.
Later, Juan Manuel Renjifo and Guiomar Caicedo de Pardo received further training at the Clodomiro Picado Institute, where they gained expertise in serpentarium management, immunization protocols, and antivenom production processes. As a result of this training, the INS manufactured its first official batch of polyvalent antivenom serum (Batch No. 4), with official production starting on June 23, 1975. Currently, the INS employs a variety of venoms (see Chapter 8). The previously produced batches No. 1, No. 2, and No. 3 were used to standardize production and quality control for Bothropic-Crotalic antivenom (AV B/C) production.
In 1975, a total of 4,020 vials of antivenom serum were produced, including 2,800 vials of polyvalent antivenom serum (SAP) and 1,220 vials of monovalent antivenom serum (SAM). From 1978 to 1990, the INS produced a total of 120,472 SAP vials and 20,334 SAM vials. SAP production peaked in the 1980s, reaching 14,811 vials in a single year.
In 1993, Law 100 of 1993, through Article 245, established the National Institute for Food and Drug Surveillance (INVIMA Spanish acronym). Its primary objective is to implement health surveillance policies and ensure the quality control of medications, biological products, food, beverages, cosmetics, medical-surgical and dental devices, homeopathic and natural products, biotechnology-derived products, and household cleaning and hygiene products. In 1994, Decree 1290 defined INVIMA's responsibilities, including enforcing policies formulated by the Colombian Ministry of Health for health surveillance and quality control of the products described in Article 245 of Law 100 of 1993, as well as other relevant regulations and recommendations from the Reviewing Commission mentioned in Article 9.
In 1995, Decree 677 partially regulated the registration and licensing regime, quality control, and health surveillance for pharmaceuticals, cosmetics, pharmaceutical preparations derived from natural resources, cleaning and hygiene products, and other household products. Additional provisions were also issued on the subject. Article 2 defines a drug as "a pharmaceutical preparation obtained from active ingredients with or without auxiliary substances, presented in a pharmaceutical form, and used for the prevention, relief, diagnosis, treatment, cure, or rehabilitation of disease." Containers, labels, and packaging are considered integral parts of the drug, as they ensure its quality, stability, and appropriate use. This classification includes serums or antivenoms.
In 2002, the INS Serum Group developed, produced, and delivered 182 vials of pan-African polyspecific serum to the WHO Collaborating Center for Antivenom Control (WHO CCCA) at the Liverpool School of Tropical Medicine, demonstrating its capability to manufacture antivenom for the African continent. During this period, the INS faced intermittent production issues, prompting the Ministry of Health to issue Resolution 122 of 2002 and declare a health emergency to ensure the supply of biologics in the country, including vaccines and antivenoms. In response, the Ministry requested the necessary resources from the PAHO/WHO Revolving Fund to reactivate antivenom production, in compliance with paragraph b) of Article 96 of Decree 677 of 1995.
In 2004, snakebite envenomations were classified as mandatory notifiable events, leading to the systematic reporting of cases in Colombia's Epidemiological Surveillance System (SIVIGILA Spanish acronym). Resolution 2934 (2004) declared a health emergency due to antivenom shortages for six months. However, this emergency declaration was extended until December 31, 2010, as the conditions necessitating it remained unresolved. This extension followed Resolution 2672 (2010) and amendments via Resolutions 613 and 5078 (2005), Resolution 2325 (2006), Resolution 2198 (2007), Resolution 2413 (2008), and Resolution 2206 (2009).
From 1991 to 2011, SAP production reached a total of 133,869 vials, while SAM production reached 19,898 vials, peaking at 16,206 SAP vials in 1998 (Figure 1). In 2005, the final production of SAM was recorded, with a total of 1,662 vials. In 2014, the INS increased its production capacity, successfully releasing approximately 3,500 vials and establishing a standard batch size of 9,500±500 SAP vials. Additionally, in 2017, the INS implemented technological upgrades to enhance and sustain production capacity, ensuring the continued availability of SAP nationwide.
In the early 2010s, the INS aimed to improve antivenom production and expand coverage of medically significant snake species. To achieve this, it developed the polyvalent coral antivenom (PAA Spanish acronym). The initial batch of antivenom, identified as 15AMP01, was released by INVIMA on October 4, 2016. This milestone in Colombian antivenom production marked the development of a polyvalent therapeutic treatment for envenomation caused by coralsnake bites (genus Micrurus), covering the four most medically important species: Micrurus dumerilii, M. isozonus,M. surinamensis (triads), and M. mipartitus (bicolor), as well as M. helleri, M. medemi, M. sangilensis,and M. obscurus by cross-neutralization [12]. Since 2016, the INS has maintained continuous PAA production, exceeding 2,000 vials per year. Additionally, it actively monitors patients receiving this antivenom as a therapy to neutralize envenomation caused by Micrurus snakes (Figure 1)
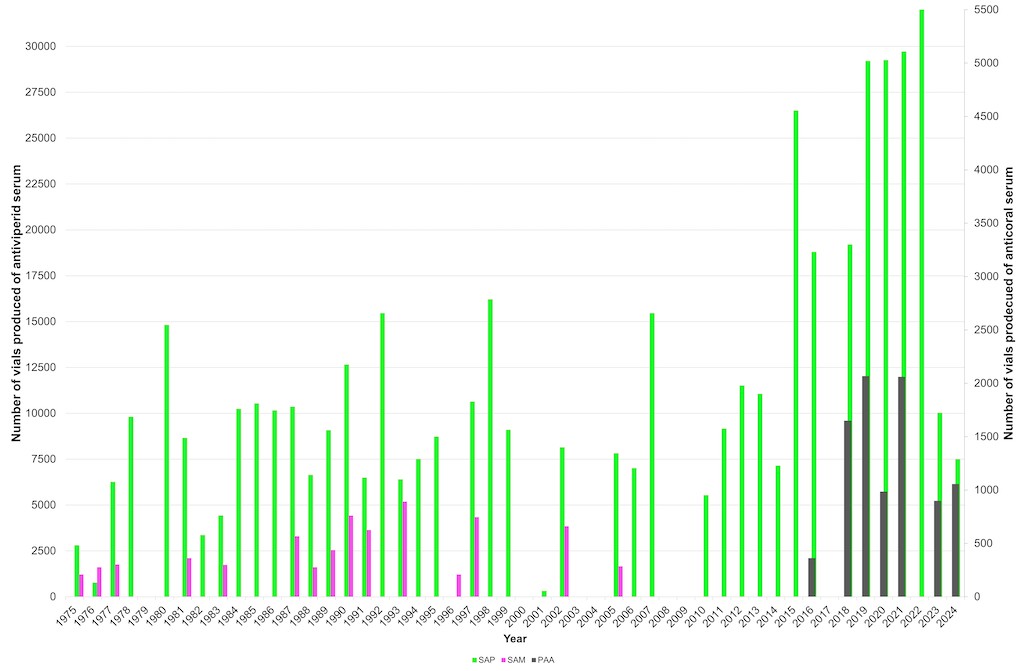
Figure 1. Historical antivenom production at the Colombian National Health Institute.
Despite the enormous efforts made by the INS to ensure the production of antivenoms, Resolutions 1300 and 1301 of April 14, 2014, declared a health emergency to prevent SAP shortages. In the same year, Decree 1375 of July 22 established the sanitary requirements for the manufacture and importation of antivenom and antilonomic sera (used as therapy for envenoming caused by caterpillars of moths of the genus Lonomia) during the declaration of a national public health emergency. Subsequently, Resolution 1209 of April 21, 2017, extended the health emergency declared by Resolutions 1300, 1301, and 1302 of 2014, which had already been extended by Resolutions 1241 of 2015, and 1478 of 2016, for twelve months.
Finally, in December 2017, the INS production plant was certified for good manufacturing practices, and the production of SAP and PAA was authorized. Additionally, INVIMA authorized the INS to manufacture pilot batches of lachetic, scorpionic, and lonomic antivenoms. Due to the scarcity of lonomic antivenom (ALP Spanish acronym), whose sole producer was the Butantan Institute in Brazil, the INS developed ALP and packaged a total of 1,663 vials between December 2017 and October 2018.
From 2012 to the present, the INS has produced a total of 182,333 vials of SAP and 7,127 vials of PAA. The maximum production per type of antivenom was achieved in 2022 with 49,931 vials of SAP and in 2019 with 2,068 vials of PAA; overall antivenom production was highest in 2022, totaling 50,525 vials, of which 594 corresponded to antilonomic antivenom (Figure 2). The plant's maximum production capacity was estimated at approximately 100,000 vials, with the qualities of SAP, which has the highest neutralization capacity among all available antivenoms in Central and South America [13].
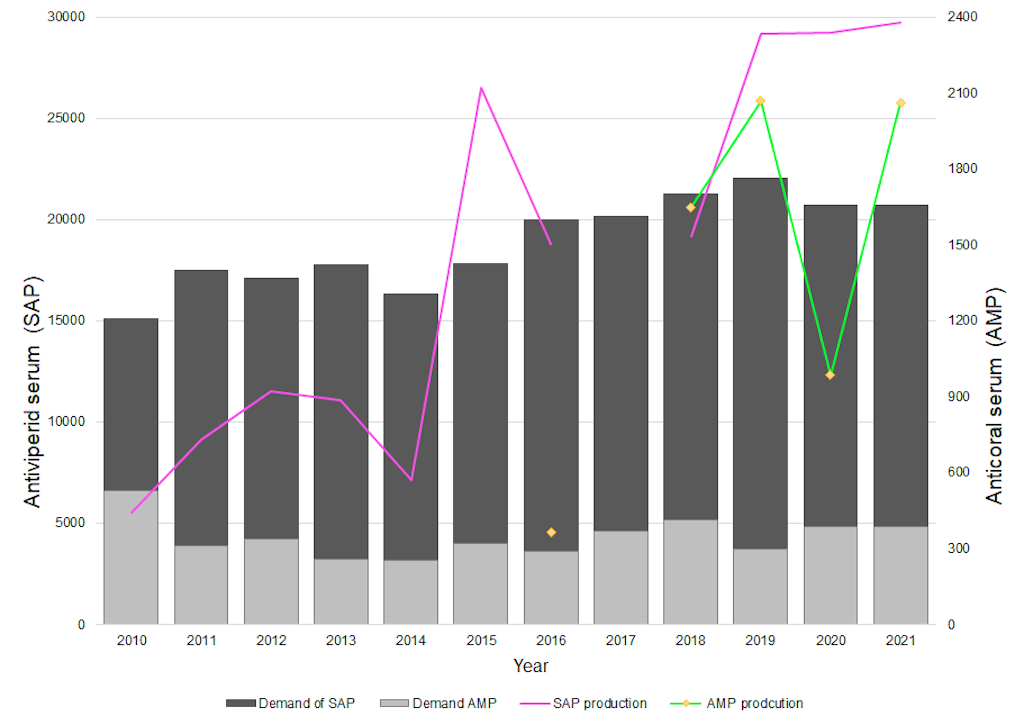
Figure 2. Antivenom production under the Good Manufacturing Practices certificate. SAP= polyvalent antiviperid serum;PAA= polyvalent anticoral antivenom.
The efficacy and quality of SAP produced by the INS have been optimized since the early 2000s by incorporating new medically important snake venoms into immunization protocols, covering all of the country's ecoregions. This enhancement increased its versatility and ensured that antivenoms retained the same antigen responsible for triggering the equine immune response. Since 2011, the INS has expanded the representativity of snake venoms from various species and biogeographical regions (see Chapters 7 and 8). This effort has resulted in a robust venom bank, which enhances the availability of venoms used for antivenom production and research. The INS maintains a stability study program for antivenom production, periodically conducting quality control tests to verify venom lethality (LD50) and evaluating the median effective dose (ED50) of all produced antivenoms to ensure their long-term stability.
The production of INS antivenoms complies with the applicable regulatory framework. Since 2010, antivenoms have been manufactured in a new hyperimmune serum plant, which holds a Good Manufacturing Practices (GMP) certification. This certification enables the INS to produce polyvalent snake antivenom (SAP), polyvalent coral snake antivenom (PAA), and polyvalent lonomic antivenom (ALP).
The polyvalent snake antivenom has held INVIMA sanitary registration number 2012M-0013350 since 2012, granted through Resolution 2012019512 (July 13, 2012), and this was renewed under number 2019M-0013350-R1 by Resolution 2019034649 (August 12, 2019). Notably, the INS manages the entire production chain, from snake collection and venom extraction to characterization and final product development, ensuring compliance with all quality standards authorized by INVIMA.
The INS guarantees antivenom production by adhering to manufacturing and quality control protocols. Compliance with all established quality specifications and parameters is verified at each stage of the production chain. Additionally, quality control includes a stability study program and active pharmacovigilance. Each manufactured batch is certified by INVIMA before commercialization (Table 1).
Table 1. Production of antivenoms carried out at the Instituto Nacional de Salud de Colombia between 1975 and 2024.
|
Historical production
|
Antivenom production
|
|
Period
|
SAP Vials
|
SAM Vials
|
PAA Vials
|
|
1975 to 1990
|
120,472
|
20,334
|
0
|
|
1991 to 2011
|
133,869
|
19,898
|
0
|
|
2012 to 2024
|
249,771
|
0
|
9,082
|
SAP: Polyvalent Antiviperid serum, PAA: Polyvalent Anticoral Antivenom, SAM: Monovalent Antiviperid serum.
To estimate the required number of vials of polyvalent antivenom serum (SAP) and polyvalent coral antivenom (PAA) produced by the INS, the minimum toxicity neutralization capacity is considered to be 7 mg/mL for Bothrops spp. venom, 1 mg/mL for Crotalus durissus venom, and 0.2 mg/mL for M. dumerilii,M. isozonus, M. surinamensis, and M. mipartitus venoms [14]. The number of vials was determined based on the criteria established by the Ministry of Health and Social Security in the "Guide for the Management of Toxicological Emergencie s" and considering the number of snakebite incidents reported in Colombia between 2010 and 2021. Figure 2 illustrates the production of SAP and PAA during the 2010–2021 period, demonstrating that the INS’s production capacity is sufficient to meet national demand.
1.3 Regulation of antivenom production
The INVIMA is a governmental entity in Colombia tasked with protecting the health of individuals and the population as a whole. INVIMA regulates and monitors the consumption of food, the use of pharmaceuticals, medical devices, and other products requiring sanitary oversight.
According to INVIMA’s regulations for medicines and biological products, antivenoms are classified as biological products [15]. INVIMA systematically monitors biological products to ensure their quality and safety standards. The National Pharmacovigilance Program under INVIMA aims to oversee the safety, efficacy, and quality of medicines during their commercialization phase after obtaining marketing authorization from INVIMA. These activities complement the control and monitoring of products throughout the production chain, minimizing health risks and impacts [11].
INVIMA authorization is required for the commercialization, importation, or manufacturing of antivenoms. This permit is granted only after compliance with all requirements established under current sanitary regulations, as validated by INVIMA [11]. The key requirements include standardization and validation of production processes and testing methods, pharmacological evaluation, manufacturing certification, sanitary registration (RS), and compliance with stability standards for the marketed antivenoms. In Colombia, biological products such as antivenoms cannot be marketed without meeting all these requirements. However, the Ministry of Health and Social Protection is responsible for ensuring the health and safety of the population. Therefore, it must take the necessary measures to prevent and control any contingencies that may affect public health, such as a potential shortage of antivenoms. As previously mentioned, in exceptional circumstances, the Ministry has declared a national health emergency on multiple occasions due to antivenom shortages.
Colombia has limited regulations governing antivenom production. Decree 821 of 2017 established the "Emergency Technical Regulation for the Obtaining of Antivenom Sanitary Registration" and adopted the "Good Manufacturing Practices (GMP) Guide" for their production. However, this decree was partially repealed by Decree 386 of 2018, which stipulates that a "scaled drawing of label drafts and packaging designs must be created, including: product name, manufacturer name, batch number, pharmaceutical form, labeled volume, specificity (neutralized venom including the common names of the animals against which the product is effective), neutralizing potency, storage conditions (including those for the reconstituted product, if applicable), description of the reconstitution process, route of administration, recommended dosage, contraindications, warnings, and expiration date." The technical annex of the decree corresponds to the "Good Manufacturing Practices (GMP) Guide for Antivenom Production".
Decree 386 of 2018 outlines the procedure for obtaining, renewing, or modifying the sanitary registration of antivenoms and includes measures to ensure their availability. Countries such as Mexico and Brazil have their own pharmacopeias, which contain specific monographs for each type of antivenom. These monographs define the quality parameters that different antivenoms must meet for production and/or commercialization within each country. In Mexico, this document is known as the "United Mexican States Pharmacopeia," issued by the Ministry of Health, while in Brazil, it is the "Brazilian Pharmacopeia," published by the National Health Surveillance Agency (ANVISA Portugues acronym).
In these countries, compliance with the monographs is mandatory, and they specify the minimum neutralizing capacity required for each type of antivenom and the venomous species it is effective against. However, Colombia lacks reference sera or venoms, and there is no pharmacological regulation establishing the minimum neutralizing capacity that antivenoms must meet or against which venoms this capacity should be tested. This legal, technical, and regulatory gap must be addressed to enhance the procedures for the sanitary registration of antivenom. Establishing such standards would ensure that all antivenoms available in the Colombian market meet the minimum neutralizing capacity required for envenomations caused by medically significant species in the country.
2. Antivenom development
Traditional antivenom production methods involve inducing antibody production in a host through serial inoculations of snake venom, taking into account its median lethal dose (LD50) [16,17,5059,63]. The horse is the most commonly used animal for immunoglobulin production due to its large blood volume, resilience, and capacity to generate antibodies against venom [18–20]. A review of studies indicates that direct inoculation of raw venom is not well tolerated by or lethal to the host [16,17,5059,63]. Therefore, adjuvants must be used during venom administration to form antigen deposits, ensuring a controlled release that preserves the immunogenicity of purified toxoids while effectively stimulating the immune response [21].
The immunization schedule varies depending on the LD50 of the venom [22], the species used for immunoglobulin production, and the protein purification technique employed [23]. It is essential to periodically analyze antibody titers at the end of the immunization cycle to assess the production of the desired immunoglobulins [5,24]. Once the target antibody titer is achieved, the horses undergo production bleeding, during which approximately eight liters of blood can be collected from each animal over a period of three to four days, depending on its physiological condition. The plasma is subsequently separated by centrifugation and precipitated using ammonium sulfate. Red blood cells and albumin are reinfused into the animal via plasmapheresis to mitigate the risk of anemia [25,26]. Historically, the production and commercialization of antivenoms in pharmaceutical form have evolved through four generations [27]. The methodology is described by Pope [28,29], which serves as the foundation for antivenom production (Figure 3).

Figure 3. Immunization procedure and immunoglobulin production. (A) Extraction of snake venom. (B–C) Inoculation of snake venom combined with adjuvants into a horse as an animal model for immunoglobulin production. (D) Catheterization of the jugular vein for blood collection. (E) Blood storage following catheterization. (F) Cell sedimentation after 12 hours of storage. (G) Mechanical separation of plasma. (H) Collection of 8 liters of hyperimmune plasma following separation. (I) Bags containing hyperimmune plasma (the raw material for antivenom production) and reconstituted cell packs in saline solution for reinfusion into the horses. (J) Plasma in quarantine, designated for processing at the manufacturing facility.
First-generation antivenoms consist of unpurified serum derived from the blood of animals hyperimmunized with venoms [30,31]. In contrast, second-generation antivenoms are composed of purified whole immunoglobulins obtained through precipitation techniques using ammonium sulfate or caprylic acid [32]. The composition of second-generation antivenoms primarily includes whole immunoglobulin G (IgG) molecules, with an approximate molecular weight of 150 kDa. It is important to note that other protein fractions, such as albumin, may also be present in these preparations [33,34].
Second-generation antivenoms are formulated with purified IgG and devoid of serum proteins. They contain trace amounts of medium and high molecular weight proteins, typically ranging between 1% and 5% [35–37]. Commercially, first- and second-generation antivenoms are referred to as sera, whereas those composed of F(ab')2 or Fab fragments are classified as fabotherapeutics [38].
Third-generation antivenoms are derived from immunoglobulin fractions of approximately 100 kDa, produced by digesting whole IgG plasma proteins with pepsin, an aspartic protease. This process cleaves the F(ab')2 fragment from the heavy chains and separates the Fc fragment from the immunoglobulins. The F(ab')2 fragment is subsequently purified through a second ammonium sulfate precipitation step [31]. These F(ab')2 fragments inhibit the interaction between the venom's active site and its receptor, thereby neutralizing the venom's effects and facilitating its elimination [39]. The molecular size is reduced as a result of Fc digestion via proteolytic cleavage, which hydrolyzes over 50% of the IgG constant regions [40]. The efficiency of this process can be influenced by the type of enzyme used and the thermocoagulation of IgG [23,26]. Additional methods for lipoprotein removal and F(ab')2 purification include ion-exchange chromatography and caprylic acid precipitation, which are occasionally employed in the purification of F(ab')2 [25].
Fourth-generation antivenoms are produced by digesting immunoglobulins with papain at neutral pH [41,42]. However, cleavage of the antibodies at the hinge region and disruption of disulfide bonds require incubation with a reducing agent, such as mercaptoethylamine, under mild conditions [43]. This results in the generation of two Fab fragments and one Fc fragment [44]. The Fc fragment can be removed from the IgG molecule through chromatographic methods [45] or by pepsin treatment, yielding purified monovalent Fab fragments [43,44]. The reduction of disulfide bonds linking the F(ab) fragments results in individual structures with intact recognition sites. Each fragment has an approximate molecular weight of 50 kDa, which enhances their clearance from the patient's body [37].
Depending on the enzymatic digestion process or the recombinant DNA technique employed, various antibody conformations can be generated to suit specific purposes [46,47]. Following antivenom production, the initial immunogenicity assessment is conducted using gel electrophoresis or western blot techniques [48,49]. An antivenom with positive immunogenicity is one that reacts with all protein fractions of the venom [50–54]. Additionally, the microbiological quality and efficacy of the product must be evaluated using the median effective dose (ED50) to determine its neutralizing capacity [55–57]. After rigorous safety and efficacy evaluations, the antivenom is packaged in vials, either in liquid or lyophilized form [25,58,59].
Antivenoms are classified as monospecific or polyspecific, depending on whether the immunization process utilizes venom from a single snake species or from multiple snake species, respectively. In both cases, the venoms are sourced from multiple populations of medically important snake species native to the region or country for which the antivenom is designed [33,60].
The production of monospecific antivenoms has largely been discontinued worldwide. However, some manufacturers continue to produce monovalent antivenoms. For instance, in Peru, the National Center for Biological Products manufactures two types of monospecific sera for snakebite treatment [7]. The injectable anticrotalic serum is used to treat envenomations caused by Crotalus durissus (South American rattlesnake), while the antilachesic serum is employed for envenomations caused by Lachesis muta (Amazonian bushmaster) [61]. In contrast, most manufacturers produce polyspecific antivenoms that neutralize the venom of viperids or elapids, such as the antiviperid and anticoral antivenoms produced in Colombia by the INS [62,63].
Experimental approaches have been proposed to develop novel antivenoms using diverse antibody formats, with the aim of creating more efficient and effective pharmaceutical treatments [64,65]. These approaches include not only the enzymatic digestion of immunoglobulins, as previously described, but also other innovative methods. For example: (1) generating monoclonal or recombinant antibodies targeting specific venom components [66]; (2) utilizing synthetic epitope chains for immunization [67]; and (3) producing recombinant antigens based on the consensus of specific toxins that are poor immunogens but can enhance existing antivenoms or lead to the development of new antivenoms with exclusive specificity [68].
Currently, international biotechnological advancements have achieved promising results in both in vivo and in vitro experiments [69,70], However, they still need to be evaluated in clinical studies. In recent research performed in Colombia, Romero-Giraldo et al. [71], cloned and expressed the most abundant phospholipase A2 (PLA2) from the venom of Micrurus dumerilii and generated IgGs against the recombinant toxin. These antibodies successfully neutralized the myotoxic activities produced by PLA2 as well as those induced by the whole venom. While these results are promising, clinical trials are still required to validate their efficacy and efficiency.
In Colombia, the use of these modern technologies for improving antivenoms as therapeutic treatments for snake envenomations remains in its nascent stages. Nevertheless, we urge researchers to explore these avenues to advance the development of next-generation antivenoms.
3. Preclinical evaluation of antivenoms
For a biological medicinal product intended for human use, such as antivenoms, to meet quality requirements, it must undergo rigorous evaluation to demonstrate its neutralizing capacity against venom toxins, as well as its effectiveness in controlling or reversing the clinical manifestations caused by envenomation. The World Health Organization (WHO) proposes methodologies for evaluating such medications. This is because the parenteral administration of antivenoms derived from animal immunoglobulins is the cornerstone of treating snakebite envenomations, which constitute a serious public health issue in tropical regions worldwide [72].
To ensure the efficacy and safety of antivenoms for human use, it is necessary to evaluate their neutralizing capacity and safety at the preclinical level. This is due to the significant variation in venom composition and the types of venoms used for immunization (see Chapters 2, 3, and 5), as well as the heterogeneity in clinical efficacy [73]. The preclinical evaluation of snake antivenoms provides valuable information for selecting appropriate antivenoms for use in various countries around the world [74,75]. The WHO recommends that the essential preclinical assays to determine the neutralizing capacity of antivenoms against venom-induced lethality are the median effective dose of the antivenom (ED50) and the median lethal dose (LD50) of the venom, respectively. However, since venoms induce other toxic activities that can also be assessed, Figure 4 outlines the most relevant steps for determining the preclinical efficacy of an antivenom during its production process.
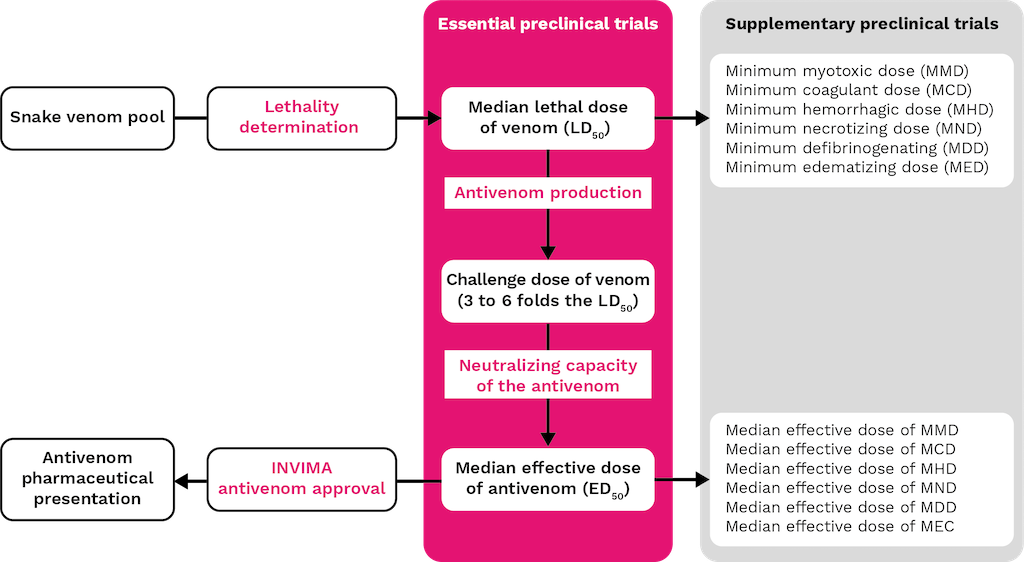
Figure 4. Preclinical evaluation process of antivenoms: Essential and supplementary tests.
3.1 Determination of the biological activities of venoms
Determination of the median lethal dose
The median lethal dose (LD50) of a venom is defined as the quantity of venom required to cause death in 50% of the test animals, typically mice. The LD50 is determined by preparing a series of venom dilutions in saline solution and administering them intraperitoneally (i.p., maximum 0.5 mL) or intravenously (i.v., maximum 0.2 mL) to groups of 4-6 mice. According to WHO guidelines [76], a sample size of five mice (of the same strain and within a defined weight range, usually 18-22 g) is recommended. Mortality is recorded at 24 hours (for intravenous injections) or 48 hours (for intraperitoneal injections). Control groups receive saline solution. The LD50 is calculated using statistical methods such as the Spearman-Karber method, Probit analysis, or other non-parametric approaches [77–80].
Determination of the median effective dose of the antivenom
The median effective dose (ED50) of an antivenom is defined as the amount of antivenom, or the venom/antivenom ratio, that ensures the survival of 50% of mice injected with a mixture of antivenom and a lethal dose of venom [76]. To perform this assay, a challenge dose of venom is first selected, typically equivalent to 3-6 times the LD50 of the venom. This dose is then mixed with varying volumes of antivenom and adjusted to a constant final volume with saline solution. The mixtures are incubated for 30 minutes at 37 °C, after which aliquots are injected into mice via the selected route (i.v. or i.p.). Survival rates are recorded based on the prepared dilutions. Two control groups are included: one injected with saline solution (negative control) and another with venom alone (positive control). The ED50 is calculated using the same statistical methods as for the LD50 and is commonly expressed as the amount of venom (in mg) neutralized per milliliter of antivenom.
In addition to these essential preclinical evaluations, which assess the ability of antivenoms to neutralize lethal effects, supplementary assays may be conducted to evaluate their efficacy against other toxic activities of venoms. These assays are optional and depend on the specific venom used, the laboratory, and the regulatory requirements of the country. For example, under Colombian regulations (Decree 386 of 2018), specific preclinical studies are required to demonstrate the neutralizing capacity of antivenoms against the target venom. The INS has developed standardized and validated biological tests to support these evaluations, including assays for myotoxicity, hemorrhage, dermal necrosis, coagulant activity, and edematizing activity, among others (Figure 4).
Determination of the minimum hemorrhagic dose and the median effective antivenom dose
The minimum hemorrhagic dose (MHD) is defined as the smallest quantity of venom that induces a hemorrhagic lesion with a diameter of 10 mm. To determine the MHD, various doses of venom are prepared in saline solution (final injection volume: 50 µL) and injected intradermally into the shaved ventral skin of mice. After 2-3 hours, the mice are ethically euthanized, and the skin is dissected to measure the diameter of the hemorrhagic lesion. A sample size of five mice per dose is recommended, with a control group receiving saline solution.
For the neutralization assay, a challenge dose of 1-5 MHD is mixed with varying doses of antivenom. The mixtures are incubated for 30 minutes at 37°C, after which 50 µL aliquots are injected intradermally. The mice are euthanized 2-3 hours later, and the skin is dissected to measure the lesion diameter. The median effective dose for MHD (MHD50) is defined as the volume of antivenom (in µL) or the venom/antivenom ratio that reduces the hemorrhagic lesion diameter by 50% compared to the control group injected with venom and saline [76].
Determination of minimum necrotizing dose and the median effective antivenom dose
The minimum necrotizing dose (MND) is defined as the smallest amount of venom that, when injected intradermally, induces necrotic lesions of 5 mm in diameter three days post-injection. The methodology is similar to that used for the MHD, except that the skin is examined three days after venom administration.
For the neutralization assay, a dose of 1-2 MND is mixed with varying doses of antivenom. The mixtures are incubated for 30 minutes at 37°C, and 50 µL aliquots are injected intradermally. Three days later, the necrotic lesion diameter is measured. The median effective dose for MND (MND50) is defined as the volume of antivenom (in µL) or the venom/antivenom ratio that reduces the necrotic lesion diameter by 50% compared to the control group [76].
Determination of minimum coagulant dose and the effective antivenom dose
The minimum coagulant dose (MCD) can be determined using either a bovine fibrinogen solution (2.0 g/L; MCD-F) or standard citrated human plasma (MCD-P). The MCD is defined as the smallest amount of venom that coagulates the fibrinogen solution or plasma within 60 seconds at 37°C. To measure the MCD, 50 µL of venom (ranging from 240 to 0.5 mg/L) is added to 0.2 mL of fibrinogen or plasma in glass coagulation tubes at 37°C. The coagulation time is recorded within 60 seconds.
For the neutralization assay, a dose of MCD-P or MCD-F is mixed with varying doses of antivenom. The mixtures are incubated for 30 minutes at 37°C, and the coagulation time is recorded. The effective dose (MCD-F100 or MCD-P100) is defined as the minimum volume of antivenom or the venom/antivenom ratio required to completely prevent coagulation within 30 minutes [76].
Determination of minimum myotoxic dose and the median effective antivenom dose
The minimum myotoxic dose (MMD) is defined as the amount of venom that increases serum or plasma creatine kinase (CK) activity to four times the level observed in control animals injected with saline solution. To determine the MMD, 50 µL of venom is injected into the right gastrocnemius muscle. After 3 hours, blood samples are collected via tail incision, and CK activity is measured using commercial diagnostic kits.
For the neutralization assay, a dose of 3 MMD is mixed with varying doses of antivenom. The mixtures are incubated for 30 minutes at 37°C and injected into the right gastrocnemius muscle. After another 3 hours, blood samples are collected, and CK activity is measured. The median effective dose for MMD (MMD50) is defined as the volume of antivenom (in µL) or the venom/antivenom ratio that reduces CK activity by 50% compared to the control group [76].
3.2 Neutralization assays
The evaluation of antivenom effectiveness is crucial for the treatment of snakebite envenomation. The WHO [76] specifies that each batch of antivenom must be tested to determine its capacity to neutralize the lethal effects of relevant venoms, using a testing method approved by the national regulatory authority of the country where the antivenom is manufactured [81]. Research on antivenoms aims to improve the treatment of snakebite envenomation through lethality assays. As previously described, these assays include the LD50, which quantifies venom lethality, and the ED50, which estimates the in vivo neutralizing potential of an antivenom [81,82].
However, several in vitro techniques have been developed to quantify venom lethality and the neutralizing capacity of corresponding antivenoms. These include immunochemical assays that evaluate the immunological interaction between antivenoms and venoms [57,82,83].Immunochemical methods, such as enzyme-linked immunosorbent assay (ELISA), can be combined with electrophoresis techniques like western blot and antivenomics studies to provide specific detection. Proteomic techniques can also be employed to identify venom proteins and their epitopes recognized by antivenoms [86], These methods allow for the quantification of antibody-antigen recognition and reduce the need for animal experimentation [83,87,88].
ELISA assays
The principle of this technique is based on the binding of soluble antigens or antibodies to an insoluble solid phase, such as the wells of a microtiter plate, which preserves the immunoreactivity of the components [59]. An antigen-antibody complex is detected using a specific enzyme-conjugated antibody, followed by a washing step. A substrate specific to the enzyme is then added, and the amount of hydrolysis (color change measured visually or spectrophotometrically) is proportional to the amount of antigen present in the test sample [87,88]. The ELISA assay is highly sensitive, capable of detecting protein levels as low as 1 to 400 ng/mL. The specificity of the assay depends on the quality of the reagents used [82,89].
Preliminary studies have shown that the ELISA system can serve as an in vitro alternative to the ED50 assay in mice, and there is a significant correlation between in vivo and in vitro ED50 values [90]. For example, the ED50 of 38 different batches of antivenom was estimated for four medically important snake venoms in Africa and compared with the in vivo ED50 [90]. ELISA plates were coated with the same venoms used for the reference curves. A significant correlation (p < 0.001) was observed between in vitro and in vivo methods when the ED50 results of the antisera were related to their optical densities [87].
To serve as a positive control, reference antivenoms (those with an established ED50 in mice) must be tested on each ELISA plate and maintained as sterile aliquots at 4°C. Generally, opacities develop before potency is lost. Lyophilized and frozen antivenoms have demonstrated long-term stability. However, standardizing and distributing reference antivenoms for each venom is impractical, which represents a significant limitation [82].
Western Blot
The term "western blot" (WB) was first coined by Dr. W. Neal Burnette [91,92]. He observed that electrophoresis facilitated the transfer of proteins from SDS-PAGE gels to nitrocellulose paper membranes and that this method performed better than chemically modified paper [92].
The western blot process begins with the electrophoretic separation of a mixture of antigens using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). This method relies on the electrotransfer of venom proteins to a nitrocellulose membrane, which is then treated with two antibodies. One of these antibodies is conjugated to an enzyme that enables the visualization of the target components.
The components bound to the nitrocellulose are accessible for interaction with antibodies or other molecules [59]. The first antibody is the antivenom, which binds to the venom proteins, while the second antibody is conjugated to an enzyme that degrades a substrate, producing a colorimetric signal that indicates the components recognized by the antivenom [59]. This technique cannot detect the recognition of structural epitopes that may be relevant, as the venom proteins in the gel are denatured. The ELISA technique is more suitable for this purpose. However, the advantage of western blot lies in its use of SDS-PAGE gels, which can detect most protein bands in the venom and provide information on the molecular weight of the recognized antigens [84,93].
3.3 Antivenomics
Variability in venom composition across different geographic populations, developmental stages, and individuals of the same species highlights the critical need for comprehensive proteomic, toxicological, and immunological studies both within and between populations (see Chapters 2, 3, and 5). These studies are essential for formulating an optimal venom mixture and developing effective antivenoms capable of neutralizing snakebites caused by any member of the target species [94].
Antivenomic assays represent a powerful tool for evaluating the ability of antivenoms to recognize specific venom components, which are often identified through mass spectrometry [52,95]. This approach utilizes a proteomics-based platform, such as antivenomic immunoreactivity analysis (Figure 5), to provide a quantitative assessment of therapeutic antibody molecules and toxin-binding molecules present in an antivenom [96]. This analysis complements in vivo and in vitro venom neutralization assays and has the potential to replace traditional immunological methods, such as ELISA and western blot, in some applications [97].
First-generation antivenomics
First-generation antivenomic techniques rely on the immunoprecipitation of antivenom-bound toxins. In this method, the whole venom is incubated with the antivenom, followed by the addition of a secondary antibody, such as horse anti-IgG [86,98]. The resulting immunoprecipitated antigen-antibody complexes contain toxins that are recognized with sufficient affinity by the antivenom antibodies. The non-immunoprecipitated toxins, which remain in the supernatant, represent venom components that are either not recognized by the antivenom or elicited the production of antibodies with very low affinity [99]. To identify these components, the reverse-phase chromatograms of the non-immunoprecipitated fraction are compared with the venom profile previously characterized through venomics [99].
Second-generation antivenomics
There are antivenom assessments based on F(ab')2 and Fab, which cannot be evaluated by immunoprecipitation [98]. An affinity chromatography column is filled with Sepharose, and a coupling buffer is added (Figure 5). After an incubation period, the molecules of IgG, F(ab')2, Fab', or Fab of an antivenom will covalently bind [100]. Next, the matrix is incubated with fixed amounts of venom, the fractions containing the unretained molecules that were not retained and those containing the proteins that were retained in the column due to their affinity to the antivenom molecules are analyzed separately by reverse phase chromatography [100]. An affinity chromatography approach is used to compare the retained and unretained fractions of the complete venom and quantify the degree to which an antivenom recognizes individual venom proteins [97,101].
The protein fraction not bound to the immobilized antibody matrix "i" is estimated using Equation 1 [101]:
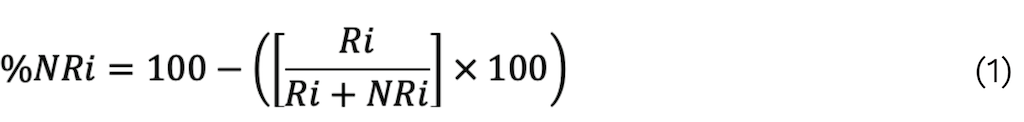
The chromatographic peak area of protein "i" in the immune retention and eluded fraction of the affinity column is presented by “Ri”. This value is used to determine the “antivenomic capacity”.
A %NRi value of ≥ 25% indicates a good result in in vivo neutralization tests [8].
Third-generation antivenomics
Third-generation antivenomics is based on the same operational principle as the second generation, which involves affinity chromatography on an immobilized antivenom matrix (Figure 5). However, instead of using a single venom concentration incubated on the antivenom matrix, a series of identical affinity columns are used. Each column is incubated with increasing amounts of venom. This modification enables determining the maximum binding capacity of each toxin in the venom to the immobilized antivenom [95,102].
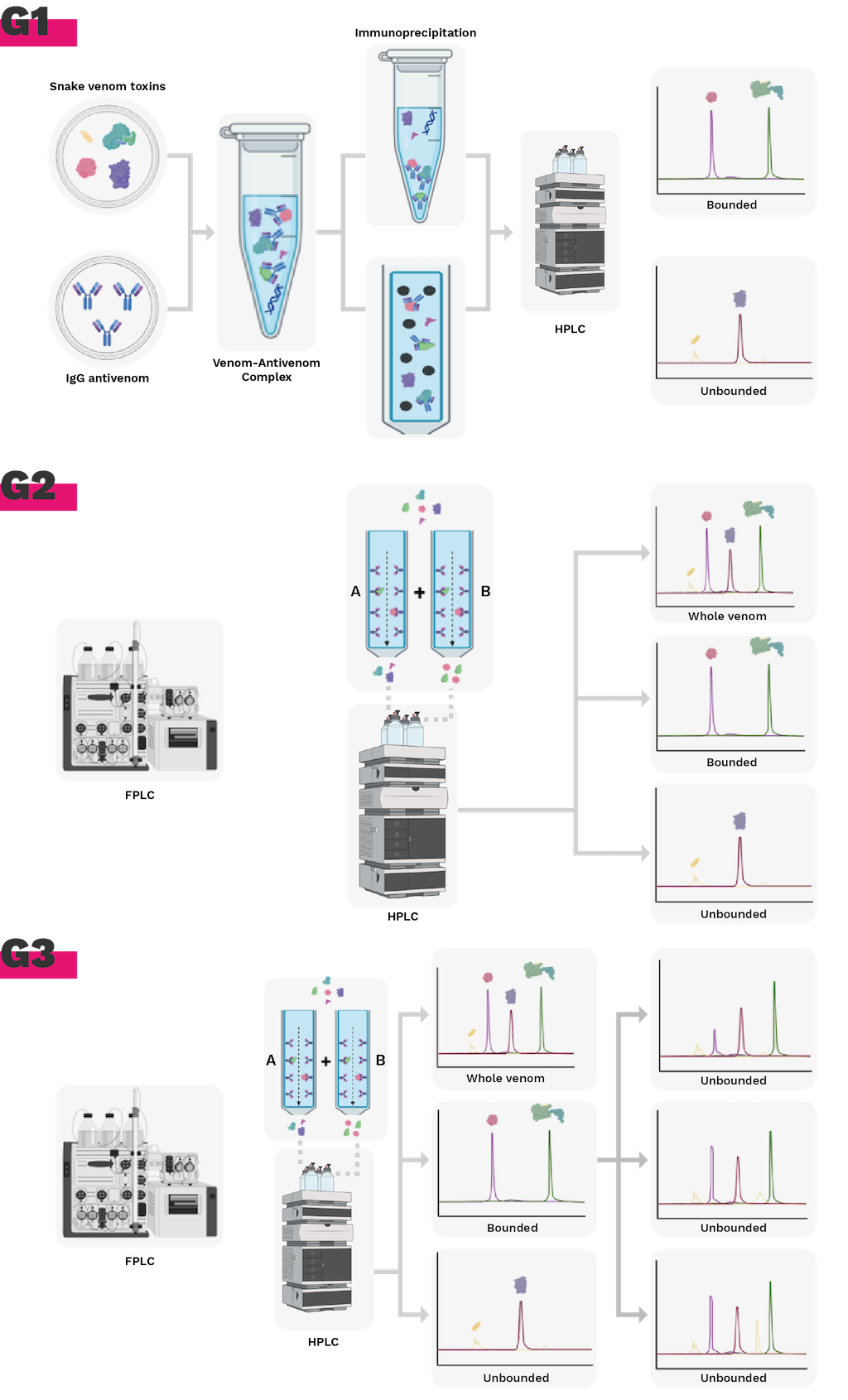
Figure 5. Evolution of immunoreactivity antivenomics methodology. G1. 1st generation antivenomics, immunoprecipitation of toxins. G2. 2nd generation antivenomics, immunoaffinity column with antivenom molecules. G3. 3rd generation antivenomics, determination of binding capacity and quantification of fractions with immunoaffinity. Figure prepared from BioRender.
However, antivenomics can only propose potential reasons for observed cross-reactivity, like identical subfamilies of crucial toxins in two venoms, one of which is employed in antivenom manufacturing. Antivenomics cannot elucidate cross-reactivity at the epitope level [98].
Despite this, the antivenoms are the only scientifically validated therapy for envenoming by snakebites. One urgent aspect that requires attention in institutions that produce antivenoms is the implementation of Good Manufacturing Practices (GMPs) to ensure the sustainability of production projects, the availability and accessibility of safe and effective antivenoms; as well as the implementation and testing of new technologies such as nano-body production (Figure 5).
4. Challenges and actions in snakebite treatment
Unlike other public health threats, snakebite envenomation is not susceptible to eradication due to the inevitable coexistence between humans and snakes, particularly in tropical regions, and the increase in encounters with these reptiles as a consequence of the expansion of human territories. Despite this reality, efforts to develop therapeutic alternatives for the management of ophidism are ongoing, and the optimization and modernization of antivenom production processes merit increased attention.
The progressive implementation of animal welfare regulations in some regions of the world poses significant challenges, including potential decreases in the number of countries with the capacity to produce antivenoms on a large scale. Consequently, clinical trial protocols using animal models need to be reviewed and updated to evaluate the cost/benefit ratio of current methods. The development and validation of alternative platforms, such as in vitro and in silico assays, designed to generate robust, reproducible, and clinically relevant results are needed. These tools will not only facilitate the transition to a more ethical paradigm in antivenom research and production but also ensure the sustainability of antivenom manufacture and universal access to this strategic biological input for public health.
Although antivenoms remain the “gold standard” therapy for the treatment of snakebite envenomation, there is a need for the production of antivenoms to be oriented towards the generation of broadly specific, affordable, safe, and effective antivenoms. This would contribute to greater homogeneity of antivenoms in all regions, reducing the cost of production and stabilizing the market for these biomolecules. The availability of good immunogens, appropriate production hosts, improved antibody purification methods, antivenom testing, including the use of alternative animal models, in vitro and in silico assays, and the establishment of appropriate distribution chains and systems to ensure that antivenoms are available in all locations, especially those closest and most prone to snakebite, are needed.